What is Retinitis Pigmentosa?
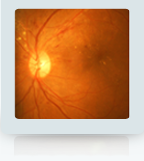
Retinitis pigmentosa (RP) refers to a group of inherited diseases causing retinal degeneration. The cell-rich retina lines the back inside wall of the eye. It is responsible for capturing images from the visual field. People with RP experience a gradual decline in their vision because photoreceptor cells (rods and cones) die.
Forms of RP and related diseases include Usher syndrome, Leber’s congenital amaurosis, rod-cone disease, Bardet-Biedl syndrome, and Refsum disease, among others.
What are the symptoms?
Symptoms depend on whether rods or cones are initially involved. In most forms of RP, rods are affected first. Because rods are concentrated in the outer portions of the retina and are triggered by dim light, their degeneration affects peripheral and night vision. When the more centrally located cones – responsible for color and sharp central vision – become involved, the loss is in color perception and central vision.
Night blindness is one of the earliest and most frequent symptoms of RP. People with mainly cone degeneration, however, first experience decreased central vision and ability to discriminate color.
RP is typically diagnosed in adolescents and young adults. It is a progressive disorder. The rate of progression and degree of visual loss varies from person to person. Most people with RP are legally blind by age 40, with a central visual field of less than 20 degrees in diameter. It is a genetic disorder and, therefore, is almost always inherited.
How is RP inherited?
An estimated 100,000 people in the U.S. have RP, mainly caused by mutated genes inherited from one or both parents. Mutated genes give the wrong instructions to photoreceptor cells, telling them to make an incorrect protein, or too little or too much protein. (Cells need the proper amount of particular proteins in order to function properly.) Many different gene mutations exist in RP. In Usher syndrome, for example, at least 14 disease-causing genes have been identified.
Genetic mutations can be passed from parent to offspring through one of three genetic inheritance patterns – autosomal recessive, autosomal dominant, or X-linked. In autosomal recessive RP, parents who carry the gene but have no symptoms themselves could have some children who are affected and others who are not. Similarly, in autosomal dominant RP, an affected parent could have affected and unaffected children. In families with X-linked RP, only males are affected; females carry the genetic trait but do not experience serious vision loss.
If a family member is diagnosed with RP, it is strongly advised that other members of the family also have an eye exam by a physician who is specially trained to detect and treat retinal degenerative disorders. Discussing inheritance patterns and family planning with a genetic counselor can also be useful.
What treatments are available?
A nutritional therapy using vitamin A (beta-carotene), vitamin C, lutein and docosahexaenoic acid (DHA) has emerged as an effective treatment for many patients; vitamin E may worsen the condition. Gene therapies are progressing through preclinical trials; technologies for delivering therapeutic agents to rod and cone cells are being studied; an implantable microchip to enhance retinal function is under development (www.FightBlindness.org).
Although not a treatment for RP, it is also important to know that low vision aids are useful for maintaining independence. Low vision specialists can make personalized recommendations for mechanical, optical, electronic, and computer-based low vision products.
What testing is available?
Genetic testing is available for RP. It helps assess the risk of passing the disorder from parent to offspring. It also helps with attaining an accurate diagnosis. A patient with an accurate diagnosis is in a better position to keep track of new findings, research developments, and treatment approaches.
However, not all RP-causing genes have been discovered. If a person chooses to get genetically tested, there is about a 50 percent chance that their disease-causing gene will be identified.